
Pioneering the Future of Chemical Discovery: How Interpretable Meta-Learning Transforms Multi-Objective Searches
In the realm of chemical research, the race to discover new molecules is both thrilling and daunting. With an estimated 1060 synthetically accessible molecules, the need for efficient and intelligent search strategies is crucial. Recent research by a team of scientists from Los Alamos National Laboratory and Georgia Institute of Technology presents a groundbreaking method that combines interpretable machine learning with traditional molecular chemistry principles. This innovative approach is designed to efficiently navigate the vast chemical landscape while addressing multiple objectives simultaneously.
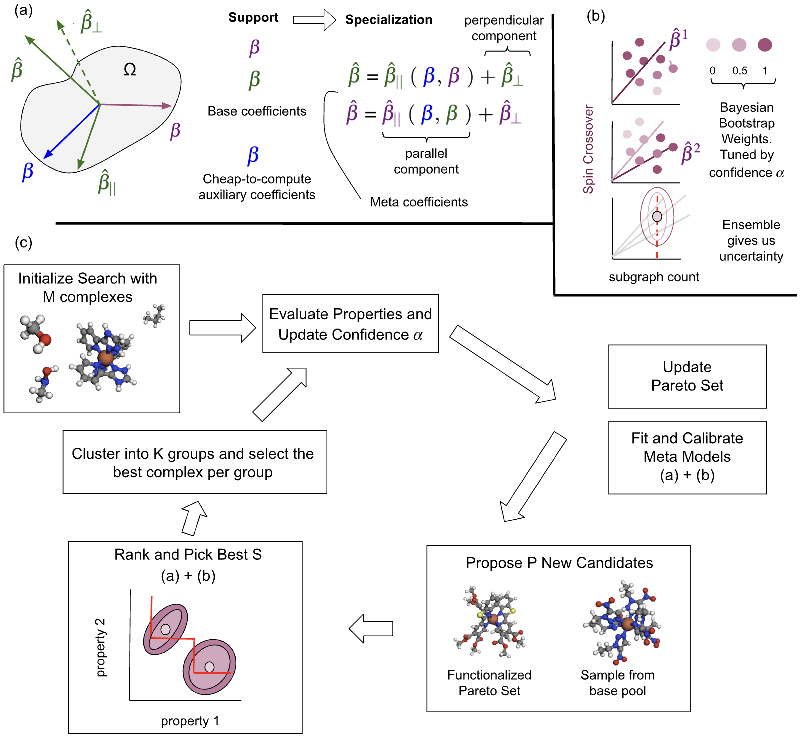
Our Approach: Meta-Learning Meets Chemical Discovery
The researchers introduced a modular pipeline that employs interpretable linear meta-learning models within an Efficient Global Optimization (EGO) framework. This unique combination allows for the simultaneous search and evaluation of multiple molecular characteristics—like stability, reactivity, and synthesizability—while ensuring each model remains interpretable and efficient to train.
Traditional deep learning methods often struggle with the computational demands of quantum-level chemistry, but the linear models used in this study excel in quick adaptability, effectively transferring knowledge across chemical tasks. By training on both chemical objectives and auxiliary properties, the meta-learned models gain valuable insights that can be quickly adapted to new, limited datasets without extensive retraining.
Key Findings: A New Benchmark for Molecular Search
In empirical evaluations, the study presented impressive results in two significant contexts: the established QM9 benchmark, which contains a diverse array of small organic molecules, and a live search for complex spin-crossover metal-organic structures. Remarkably, the meta-learning approach showed a remarkable reduction in the number of iterations needed to converge towards optimum solutions, outperforming traditional search methods by estimates nearing two orders of magnitude.
In the large-scale search for spin-crossover complexes, the researchers found that their framework outperformed standard baselines in approximately 78% of cases. The adoption of dynamic confidence tuning—an innovative method for adapting the balance between exploration (searching uncertain areas) and exploitation (focusing on known valuable areas)—further enhanced the efficacy of the search process.
Combatting Challenges with Enhanced Uncertainty Management
One of the salient challenges in multi-objective molecular discovery lies in predicting uncertainties during active searches. The research team tackled this issue by introducing a Bayesian bootstrapping method that quantifies uncertainties more accurately. By recalibrating exploration-exploitation boundaries dynamically, the methodology ensures optimal sampling of candidate molecules, even in complex and evolving distributions—essential for practical applications in drug design and materials science.
Conclusions: Implications for Future Research and Applications
This pioneering work not only provides a more effective methodology for chemical discovery but also opens the door for future advancements in the field. The modularity of the pipeline means that components like the meta-learning surrogate and uncertainty quantification methods can be independently refined or replaced. By improving the efficiency of molecular searches and providing interpretable outcomes, these innovations promise to accelerate developments in various domains, from pharmaceuticals to advanced materials.
As the journey into the uncharted territories of synthetic chemistry continues, this fusion of artificial intelligence with chemical science demonstrates a vital step towards unlocking new possibilities and pushing the boundaries of what is achievable in molecular discovery.
Authors: Antonio Varagnolo, Yulia Pimonova, Michael G Taylor, Raphaël Pestourie, Nicholas Lubbers





